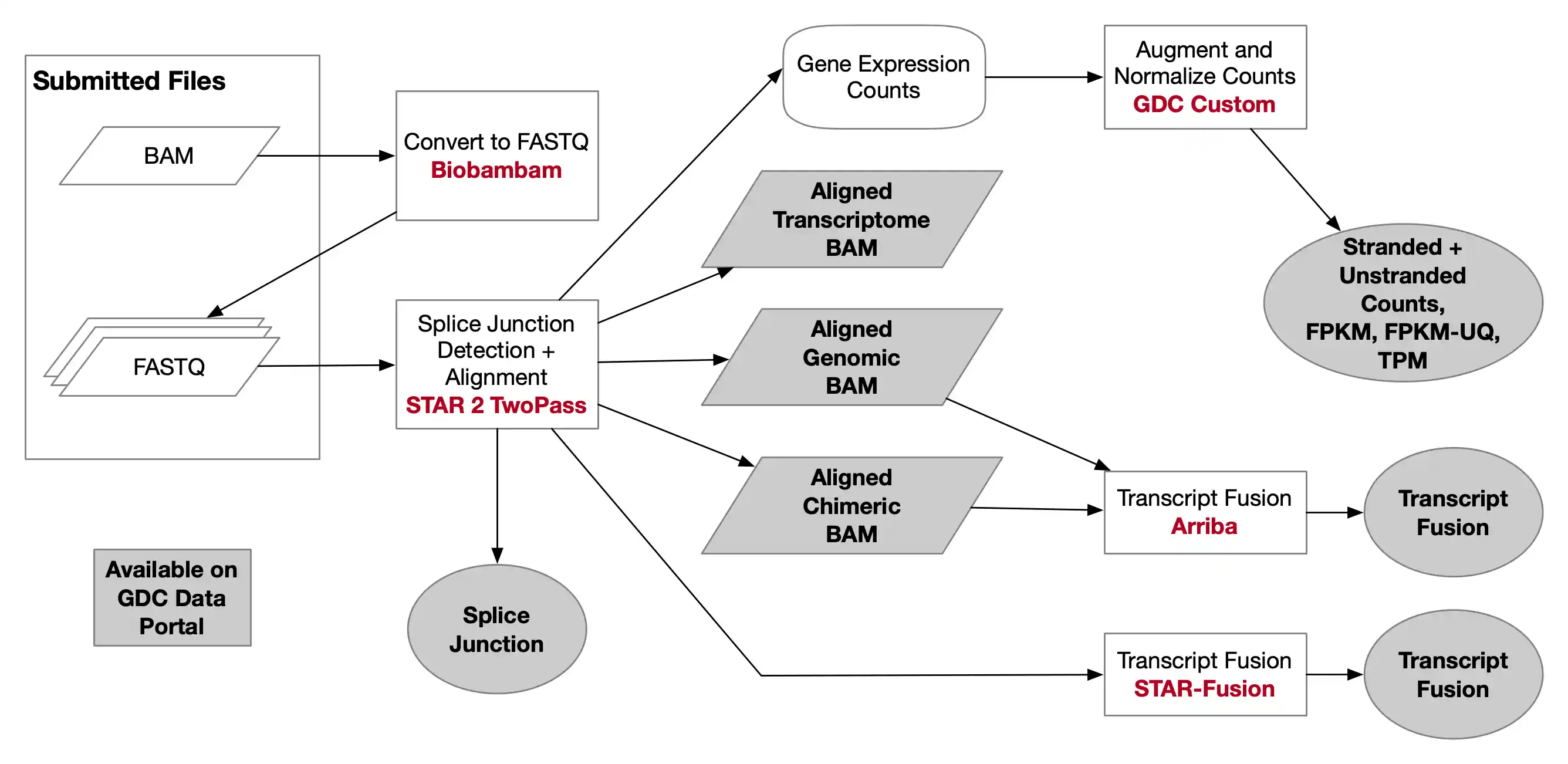
流程:https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
第一步 安装必要的软件
- conda create -n star -c bioconda star -y
- conda activate star
- conda install -c bioconda samtools -y
说明书:https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf
第二步 建立基因组索引
地址:https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files
如果要自己构建,可以使用 zcat R1.fq.gz head 来查看reads长度,选用reads长度减1(149)作为 --sjdbOverhang 比默认的100要好,但是说明里认为绝大多数情况下100和理想值差不多
https://www.gencodegenes.org/human/release_36.html
1
2
3
4
5
6
7
| STAR \
--runMode genomeGenerate \
--genomeDir index_150 \
--genomeFastaFiles GRCh38.primary_assembly.genome.fa \
--sjdbOverhang 149 \
--sjdbGTFfile gencode.v36.primary_assembly.annotation.gtf \
--runThreadN 8
|
第三步 第一次对比
CleanData存放格式
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
| #!/bin/sh
#设置CleanData存放目录
CLEAN=/home/jovyan/upload/yy_zhang(备份)/RNA-seq/Cleandata
#设置这一步的输出目录 (确保目录存在)
WORK=/home/jovyan/upload/zl_liu/star_data/yyz_01/output
#设置index目录
INDEX=/home/jovyan/upload/zl_liu/star/index_150
#设置参考文件目录
Reference=/home/jovyan/upload/zl_liu/star
echo $CLEAN", "$WORK", "$INDEX
export CDIR=$(basename `pwd`)
echo $CDIR
echo $CLEAN
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $SAMPLE
r1=${SAMPLE}"_R1.fq.gz"
r2=${SAMPLE}"_R2.fq.gz"
echo $r1
echo $r2
mkdir $WORK"/"$SAMPLE
cd $WORK"/"$SAMPLE
STAR \
--genomeDir $INDEX \
--readFilesIn $CLEAN/$SAMPLE/$r1","$CLEAN/$SAMPLE/$r2 \
--runThreadN 4 \
--outFilterMultimapScoreRange 1 \
--outFilterMultimapNmax 20 \
--outFilterMismatchNmax 10 \
--alignIntronMax 500000 \
--alignMatesGapMax 1000000 \
--sjdbScore 2 \
--alignSJDBoverhangMin 1 \
--genomeLoad LoadAndRemove \
--readFilesCommand zcat \
--outFilterMatchNminOverLread 0.33 \
--outFilterScoreMinOverLread 0.33 \
--sjdbOverhang 149 \
--outSAMstrandField intronMotif \
--outSAMtype None \
--outSAMmode None \
done
|
第四步 建立中间索引
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
| #!/bin/sh
#设置CleanData存放目录
CLEAN=/home/jovyan/upload/yy_zhang(备份)/RNA-seq/Cleandata
#设置这一步的输出目录 (确保目录存在)
WORK=/home/jovyan/upload/zl_liu/star_data/yyz_01/output
#设置index目录
INDEX=/home/jovyan/upload/zl_liu/star/index_150
#设置参考文件目录
Reference=/home/jovyan/upload/zl_liu/star
echo $CLEAN", "$WORK", "$INDEX
export CDIR=$(basename `pwd`)
echo $CDIR
echo $CLEAN
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $SAMPLE
r1=${SAMPLE}"_R1.fq.gz"
r2=${SAMPLE}"_R2.fq.gz"
echo $r1
echo $r2
cd $WORK"/"$SAMPLE
mkdir IIG
STAR \
--runMode genomeGenerate \
--genomeDir IIG \
--genomeFastaFiles $Reference"/GRCh38.primary_assembly.genome.fa" \
--sjdbOverhang 149 \
--runThreadN 4 \
--sjdbFileChrStartEnd SJ.out.tab \
done
|
第五步 第二次对比
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
| #!/bin/sh
#设置CleanData存放目录
CLEAN=/home/jovyan/upload/yy_zhang(备份)/RNA-seq/Cleandata
#设置这一步的输出目录 (确保目录存在)
WORK=/home/jovyan/upload/zl_liu/star_data/yyz_01/output
#设置index目录
INDEX=/home/jovyan/upload/zl_liu/star/index_150
#设置参考文件目录
Reference=/home/jovyan/upload/zl_liu/star
echo $CLEAN", "$WORK", "$INDEX
export CDIR=$(basename `pwd`)
echo $CDIR
echo $CLEAN
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $SAMPLE
r1=${SAMPLE}"_R1.fq.gz"
r2=${SAMPLE}"_R2.fq.gz"
echo $r1
echo $r2
cd $WORK"/"$SAMPLE"/IIG"
ln -s $INDEX/exonGeTrInfo.tab .
ln -s $INDEX/exonInfo.tab .
ln -s $INDEX/geneInfo.tab .
ln -s $INDEX/sjdbList.fromGTF.out.tab .
ln -s $INDEX/transcriptInfo.tab .
mkdir $WORK"/"$SAMPLE"/Res"
cd $WORK"/"$SAMPLE"/Res"
STAR \
--genomeDir ../IIG \
--readFilesIn $CLEAN/$SAMPLE/$r1","$CLEAN/$SAMPLE/$r2 \
--runThreadN 4 \
--quantMode TranscriptomeSAM GeneCounts \
--outFilterMultimapScoreRange 1 \
--outFilterMultimapNmax 20 \
--outFilterMismatchNmax 10 \
--alignIntronMax 500000 \
--alignMatesGapMax 1000000 \
--sjdbScore 2 \
--alignSJDBoverhangMin 1 \
--genomeLoad LoadAndRemove \
--limitBAMsortRAM 35000000000 \
--readFilesCommand zcat \
--outFilterMatchNminOverLread 0.33 \
--outFilterScoreMinOverLread 0.33 \
--sjdbOverhang 149 \
--outSAMstrandField intronMotif \
--outSAMattributes NH HI NM MD AS XS \
--outSAMunmapped Within \
--outSAMtype BAM SortedByCoordinate \
--outSAMheaderHD @HD VN:1.4 \
--outSAMattrRGline ID:sample SM:sample PL:ILLUMINA
done
|
第六步 组装Counts文件
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
WORK='/home/jovyan/upload/zl_liu/star_data/yyz_01/output'
Reference='/home/jovyan/upload/zl_liu/star'
Reference = file.path(Reference, 'index_150', 'geneInfo.tab')
file_list <- list.dirs(WORK, recursive=F, full.names = F)
geneN <- read.table(file = Reference, sep = '\t', skip = 1)
colnames(geneN) <- c('ID', 'symbol', 'type')
for(sample in file_list){
test_tab <- read.table(file = file.path(WORK, sample, 'Res', 'ReadsPerGene.out.tab') , sep = '\t', header = F)
test_tab <- test_tab[-c(1:4), ]
geneN[sample] <- test_tab[2]
}
write.csv(x = geneN, file = 'yyz_01.csv')
|
1
2
3
4
5
6
7
8
9
10
| f_name_dedup <- function(lc_exp, rowN = 1){
res <- lc_exp[-rowN]
lc_tmp = by(res,
lc_exp[[rowN]],
function(x) rownames(x)[which.max(rowMeans(x))])
lc_probes = as.character(lc_tmp)
res = lc_exp[rownames(res) %in% lc_probes,]
rownames(res) <- res[[rowN]]
res[-rowN]
}
|
第七步 删除临时文件
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
| #!/bin/sh
#设置CleanData存放目录
CLEAN=/home/jovyan/upload/yy_zhang(备份)/RNA-seq/Cleandata
#设置这一步的输出目录 (确保目录存在)
WORK=/home/jovyan/upload/zl_liu/star_data/yyz_01/output
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $SAMPLE
rm -rf $WORK"/"$SAMPLE"/IIG"
done
|