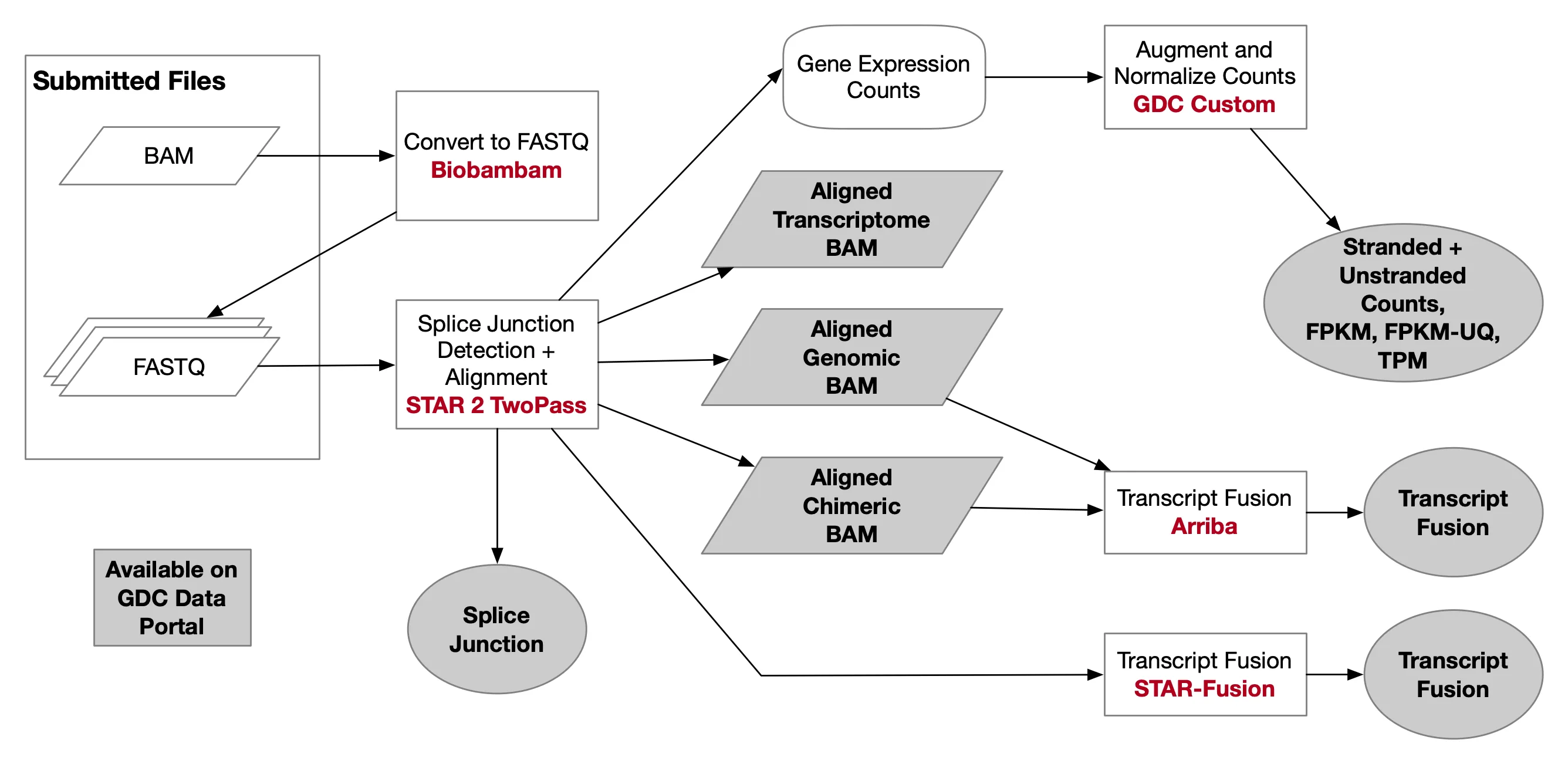
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
| library(DESeq2)
count_all <- read.csv("liver.csv",header=TRUE,row.names=1)
count_all
cts_b <- count_all[ ,c(-1,-2,-3)]
rownames(cts_b) <- count_all$ID
cts_bb <- cts_b
cts_b <- cts_bb[,c('XL07', 'XL08', 'XL09', 'XL10', 'XL11', 'XL12')]
keep <- rowSums(cts_b) > 10
cts_b[keep,]
conditions <- factor(c(rep("Control",3), rep("XL",3)))
colData_b <- data.frame(row.names = colnames(cts_b), conditions)
colData_b
dds <- DESeqDataSetFromMatrix(countData = cts_b[keep,],
colData = colData_b,
design = ~ conditions)
dds <- DESeq(dds)
res <- results(dds)
rres <- cbind(count_all[keep,c(1,2,3)], data.frame(res))
write.csv(rres, file='XL101112vs789_DESeq2.csv')
rres
|