先从TCGA的数据中提取一份标准的基因长度,作为正确结果的标准,然后开始提取工作。
Shell预处理
1
2
3
4
5
6
7
8
9
| #!/bin/bash
GFT=/home/jovyan/upload/zl_liu/star/gencode.v36.primary_assembly.annotation.gtf
awk '{if ($3=="exon") print $0}' $GFT > exon.gtf
sed -i 's/"//g' exon.gtf
awk 'BEGIN{FS="\t ;";OFS="\t"}{print $1,$3,$4,$5,$7,$10,$5-$4+1}' exon.gtf > exon.txt
|
R进行组装
1
2
3
4
5
6
7
8
| library(tidyverse)
df <- read.table('exon.txt', header = F, sep = '\t', allowEscapes = T, quote = '')
df
df <- df %>% group_by(V6)
df <- df %>% summarise(length=sum(V7), chr=unique(V1), strand=unique(V5), start=min(V3), end=max(V4))
df[['width']] = df[['end']] - df[['start']]
df
write.csv(df,'gene_length_human.csv')
|
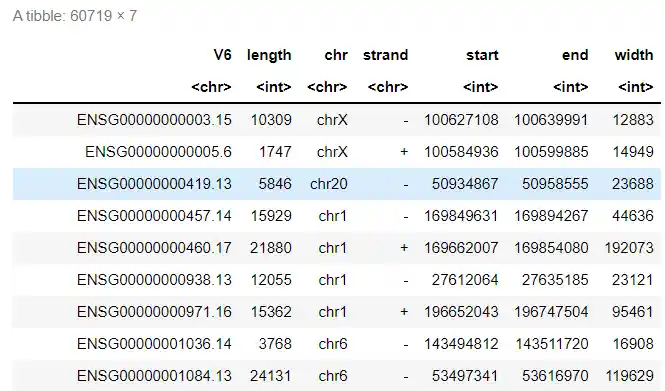
和TCGA结果一致,说明结果可信,且length理论上比width更好